Accuracy of occurrence and abundance estimates from insect metabarcoding
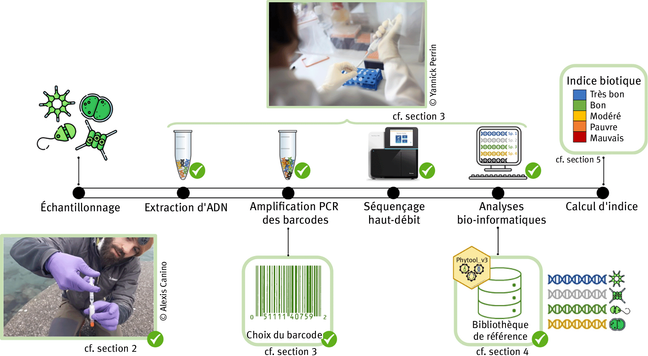
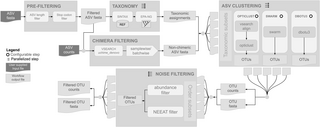
DNA metabarcoding - high-throughput sequencing of barcode regions from bulk samples - has become a key tool for insect biodiversity assessment. Yet, how methodological choices affect the accuracy of metabarcoding data remains insufficiently explored. In this paper, we ask: (1) How does the lysis method (non-destructive lysis vs. destructive homogenization) affect community recovery? (2) How comprehensively does metabarcoding capture species richness? (3) To what extent can spike-ins improve abundance estimates? (4) How accurately can species abundances be estimated? We evaluated the accuracy of insect metabarcoding using 4,749 bulk samples from a large-scale biodiversity survey subjected to mild lysis. Of these samples, 856 were also homogenized, allowing a systematic comparison of the effect of alternative treatments. To potentially improve abundance estimates, we added six biological spike-ins (i.e., foreign insects) to all samples, and two synthetic spike-ins (artificial DNA fragments) to the homogenization treatment. In addition, we established the contents of 15 samples by individually barcoding all specimens, enabling direct assessment of occurrence and abundance estimates. Our results revealed consistent differences between destructive and non-destructive treatments. While both methods reliably detected the majority of species, small and soft-bodied taxa were more often recovered after mild lysis than after homogenization, while the reverse was true for heavily sclerotized, hairy, and large taxa. Using biological spike-ins for calibration reduced the variance in read numbers per specimen considerably, especially in homogenized samples, while synthetic spike-ins were less effective. In a Bayesian analysis, where species data were matched to the best-fitting spike-in calibration curve, accurate abundance estimates (+/-1 individual) were obtained for 72.9% of species occurrences. Our results show that it is possible to obtain reasonably accurate abundance estimates from metabarcoding data, and that mild lysis and homogenization result in different taxon-specific biases in terms of occurrence data, with neither method outperforming the other. Accuracy is improved by homogenization rather than mild lysis of samples, and by the use of biological rather than synthetic spike-ins. Together, these findings provide a major step towards robust, quantitative biodiversity monitoring using DNA-metabarcoding.
### Competing Interest Statement
The authors have declared no competing interest.
Knut and Alice Wallenberg Foundation, https://ror.org/004hzzk67, 2017.088
Swedish Research Council, https://ror.org/03zttf063, 2021-04830
National Science Centre, https://ror.org/03ha2q922, 2018/31/B/NZ8/01158, 2021/43/B/NZ8/03376