As it happens, htslib has an "AI" policy
"Conformance with the software licence" is incompatible IMHO, all LLM generations should be GPL licence compatible
https://github.com/samtools/htslib/blob/develop/CONTRIBUTING.md#ai-policy
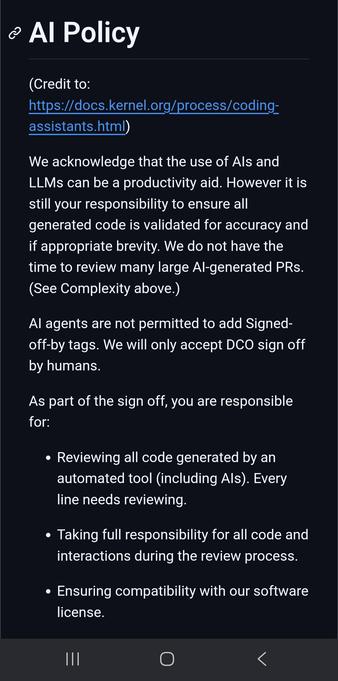
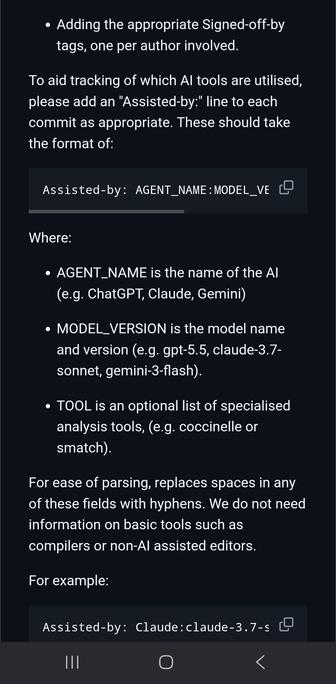
As it happens, htslib has an "AI" policy
"Conformance with the software licence" is incompatible IMHO, all LLM generations should be GPL licence compatible
https://github.com/samtools/htslib/blob/develop/CONTRIBUTING.md#ai-policy
Have you ever touched Urtica dioica? It is a memorable experience.
Beyond its sting, this plant has important medicinal properties. To better understand the genetics underlying key traits, we sequenced and analyzed its genome.
Read more here:
https://doi.org/10.64898/2026.05.15.725508

🇯🇵 🧬 🇯🇵 🧬 🇯🇵 🧬
Decoding triancestral origins, archaic introgression, and natural selection in the Japanese population by whole-genome sequencing
"We showcased potential clinical usages of JEWEL and examined the genetic legacy of Neanderthals and Denisovans in the Japanese and investigated their associations with various phenotypes, which constitutes the largest non-European analysis to date."
Xiaoxi Liu et al., Decoding triancestral origins, archaic introgression, and natural selection in the Japanese population by whole-genome sequencing. Sci. Adv. 10, eadi8419(2024). DOI: https://doi.org/10.1126/sciadv.adi8419.
#OpenAccess #OA #Research #Article #Science #Genetics #Genomics #Japan #Academia
Low-hanging fruit: everyone sees it, few actually pick it up 🍎🍇🍉🍓🫐🍒
Our latest preprint explores dark pigmentation in blackberry, a plant characterized by low-hanging fruit. The combination of genomics and transcriptomics reveals insights into the genetics of pigment biosynthesis. High levels of cyanidin-3-O-glucoside were identified in blackberries, which may explain their dark coloration.
Read more: https://doi.org/10.64898/2026.05.05.723051
#PlantSciences #Pigments #Fruits #Genomics #RNAseq
@PuckerLab

🧬🖥️ How do we keep #AI in #genomics safe?
A new paper coauthored by #RKI researchers explores risks such as data bias, quality issues, and #DualUse potential in #GenAI. It highlights how these risks emerge along the innovation pipeline and outlines strategies for safer, responsible use.

