It was a great pleasure to contribute to this work by Jemma Fendley, @mrcmlr and Boris Shraiman on pan-genomes, linkage, and recombination in phage genomes.
We analyzed data collected by the fantastic SEA-PHAGES program in https://phagesdb.org.
[1/6]
https://www.biorxiv.org/content/10.1101/2025.08.12.669904v1
The Actinobacteriophage Database | Home
Thrilled to announce that our preprint (together with fantastic @richardneher and Liam Shaw) has been officially published in Molecular Biology and Evolution! 🎉
If you're curious about how rapidly the genome of E. coli evolves (picking up new genes, rearranging, breaking synteny...) give it a look! 🧬
https://academic.oup.com/mbe/advance-article/doi/10.1093/molbev/msae272/7942412
A summary thread 🧵 https://mstdn.science/@mrcmlr/112772499102225806
A great article by Jon Cohen covering @pathoplexus & our values, with quotes from Exec Board member Anderson Brito & the community, @eddieholmes &
Gustavo Palacios!
And a great excuse to deep-dive into some of the people behind Pathoplexus! 👩🏻🔬👨🏾🔬👨🏿🔬👩🏼🔬
1/9
https://mstdn.science/@pathoplexus/113050084920833583
From Science Magazine @sciencemagazine: “We believe that building trust through transparency is essential for encouraging broader participation in data sharing,” says Pathoplexus co-founder Anderson Fernandes de Brito
https://science.org/content/article/new-scientist-run-virus-database-vows-be-transparently-run-and-simple-use
#Pathoplexus #OpenScience
[13/13]
To explain the total structural diversity of the dataset, more than 2000 distinct structural variations must have happened in its short evolutionary history, corresponding to an average rate of one every 3 mutations on the (recombination-filtered) core-genome. This is a remarkably high rate! We are curious to check in follow-up works whether these rates and patterns are specific to ST131, or are more general and can be found in other sequence types or even microbial species.
[12/13]
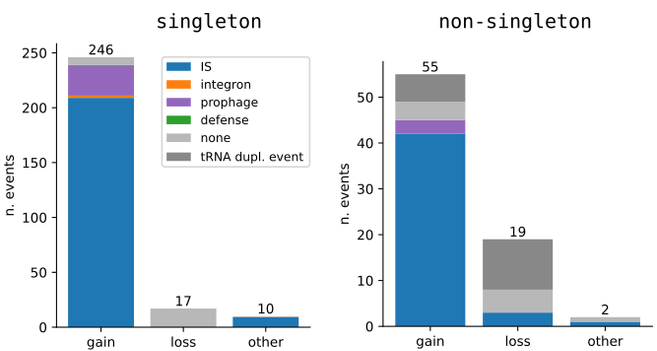
Most of the IS integrations disrupt genes, and such structural gains would be interpreted as loss events in gene-based analyses. However, this happens less than expected by chance given the high fraction of coding sequence in these genomes, indicating that roughly 2/3 of these integrations have already been removed by purifying selection.
[11/13]
In binary junctions the vast majority of events are gains, often corresponding to an insertion sequence (IS) or prophage integrating in an otherwise conserved region of the genome. This corresponds to a rough rate of one of these events every 20 mutations on the (recombination-filtered) core-genome.
[10/13]
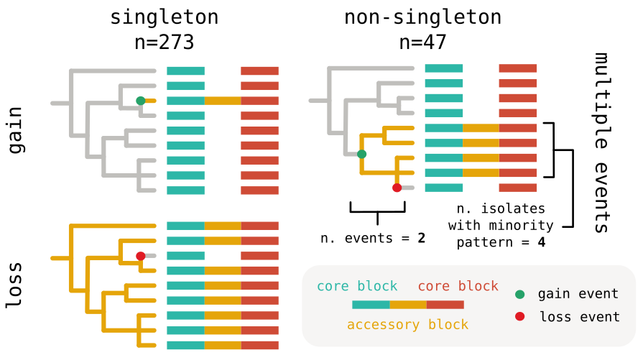
For binary junctions we can go even further: depending on their content and pattern of variation along the tree, they can be associated with gain or loss events.
In particular singleton junctions correspond to events on terminal branches of the tree, while non-singleton junctions can in principle be associated also to events on internal branches.
[9/13]
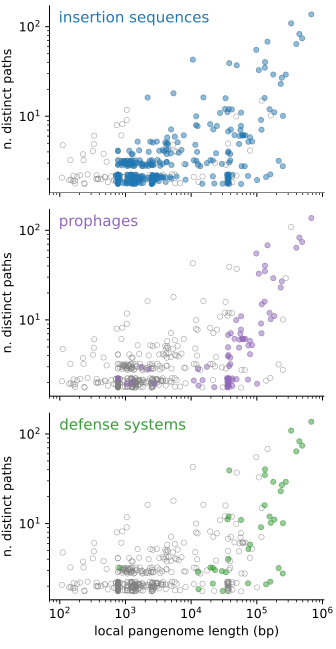
By looking at the content of the junctions, we find that the two peaks in binary junctions are explained by the movement of insertion sequences and prophages respectively, while hotspots are very flexible regions, rich in mobile genetic elements and defense systems.
[8/13]
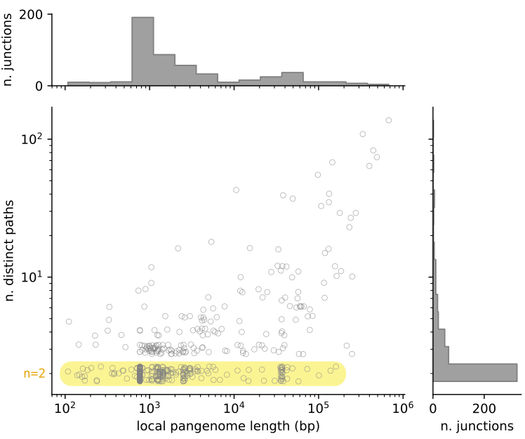
If we scatter-plot these two quantities for all of the 519 junctions in the dataset, we find that the majority of junctions are binary, i.e. they contain only two possible distinct paths, of which one is often empty. Their length distribution is bimodal, with a peak around 1kbp and another around 30-40 kbp. On the other end of the spectrum we find hotspots, regions with tens to hundreds of different distinct paths, and a total accessory genome content of tens to hundreds of kbp in length.