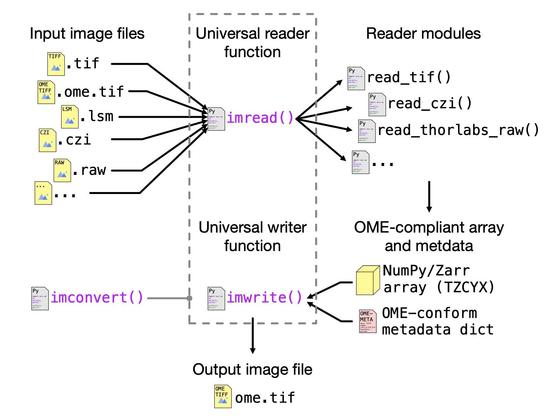
If you work with #microscopy images 🧠🔬 in #Python 🐍👨💻, you may know the situation: Different file formats, readers, axis conventions, metadata structures... At least I found this increasingly frustrating. So I built #OMIO (Open Microscopy Image I/O), a unified microscopy image reader & writer for #Python:
🌍 https://omio.readthedocs.io/
🧵1/8