Great news!
Our latest #OpenAccess paper has been published by @MicrobioSoc
📌Rapid identification and subsequent contextualization of an outbreak of methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit using @nanopore sequencing


Rapid identification and subsequent contextualization of an outbreak of methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit using nanopore sequencing
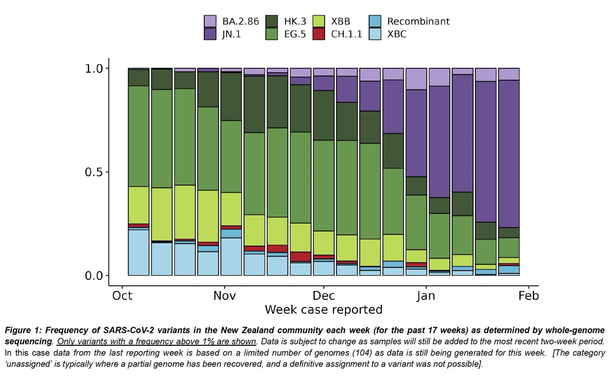
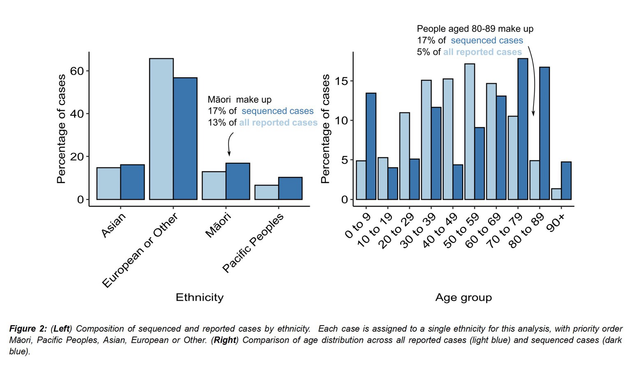
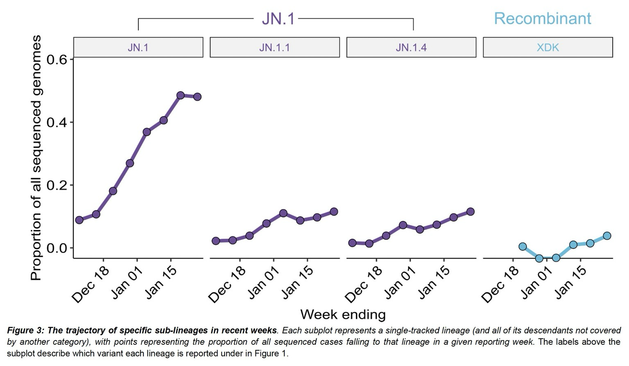
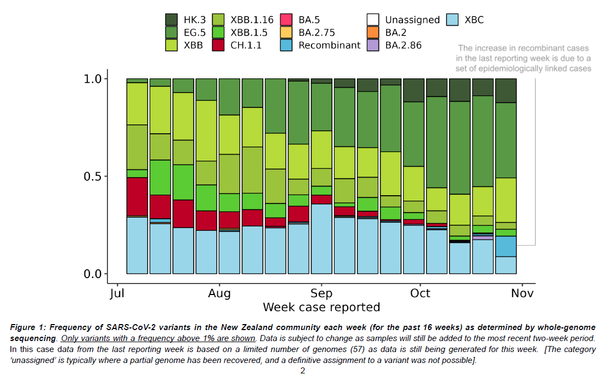
Outbreaks of methicillin-resistant Staphylococcus aureus (MRSA) are well described in the neonatal intensive care unit (NICU) setting. Genomics has revolutionized the investigation of such outbreaks; however, to date, this has largely been completed retrospectively and has typically relied on short-read platforms. In 2022, our laboratory established a prospective genomic surveillance system using Oxford Nanopore Technologies sequencing for rapid outbreak detection. Herein, using this system, we describe the detection and control of an outbreak of sequence-type (ST)97 MRSA in our NICU. The outbreak was identified 13 days after the first MRSA-positive culture and at a point where there were only two known cases. Ward screening rapidly defined the extent of the outbreak, with six other infants found to be colonized. There was minimal transmission once the outbreak had been detected and appropriate infection control measures had been instituted; only two further ST97 cases were detected, along with three unrelated non-ST97 MRSA cases. To contextualize the outbreak, core-genome single-nucleotide variants were identified for phylogenetic analysis after de novo assembly of nanopore data. Comparisons with global (n=45) and national surveillance (n=35) ST97 genomes revealed the stepwise evolution of methicillin resistance within this ST97 subset. A distinct cluster comprising nine of the ten ST97-IVa genomes from the NICU was identified, with strains from 2020 to 2022 national surveillance serving as outgroups to this cluster. One ST97-IVa genome presumed to be part of the outbreak formed an outgroup and was retrospectively excluded. A second phylogeny was created using Illumina sequencing, which considerably reduced the branch lengths of the NICU isolates on the phylogenetic tree. However, the overall tree topology and conclusions were unchanged, with the exception of the NICU outbreak cluster, where differences in branch lengths were observed. This analysis demonstrated the ability of a nanopore-only prospective genomic surveillance system to rapidly identify and contextualize an outbreak of MRSA in a NICU.