#Duckdb #htslib #Genomics #Bioinformatics #RStats
duckths: Read HTS (VCF/BCF/BAM/CRAM/FASTA/FASTQ/GTF/GFF) files in DuckDB via htslib
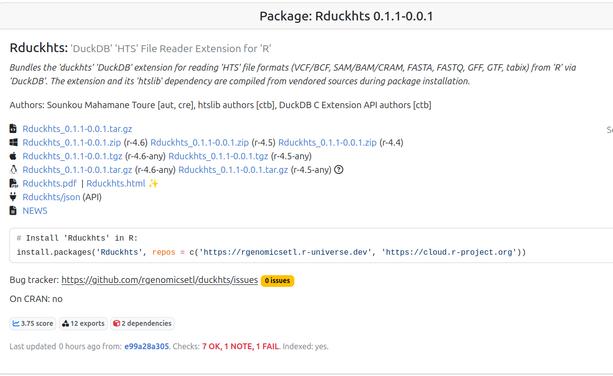
Rduckhts: 'DuckDB' High Throughput Sequencing File Formats Reader Extension
genomic.social/@bioinfhotep...
SMT (@[email protected]...
SMT (@[email protected]...

SMT (@[email protected])
Attached: 4 images #Genomics #Bioinformatics Release of duckhts: #htslib based #Duckdb Extension for High Throughput Sequencing File Formats https://duckdb.org/community_extensions/extensions/duckhts Allied Self contained #Rstats package Rduckhts : https://rgenomicsetl.r-universe.dev/Rduckhts For now the lifecycle of the APIs (Duckdb C API extension and R package) are "experimental". They are mostly targeted to some ETL use cases, but will add some some useful scalar and aggregation functions including Heng Li's C Genomic ranges Feedback and testing welcome !