https://seqera.io/multiqc_config_wizard
#MultiQC Configuration Wizard
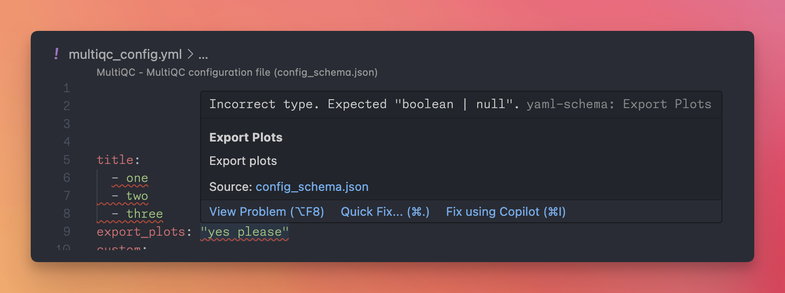
"this tool helps you create a custom multiqc_config.yaml configuration file. Edit the form on the left, or paste an existing config into the editor on the right. Each option is checked against the schema; status badges and filters let you narrow in on what's set, broken, or unrecognised. "