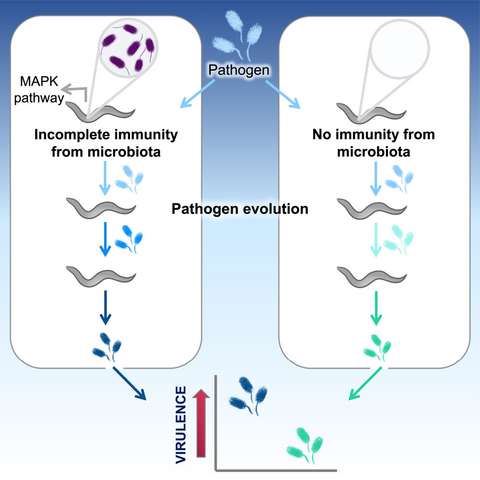
New article from postdoc Kim Hoang and Kayla King (Oxford/ U British Columbia) looking at how incomplete immunity to a pathogen provided by a competing "commensal" species can have the unintended effect of selecting for more virulent pathogens. Cool experimental evolution!
https://www.sciencedirect.com/science/article/pii/S096098222400157XExeter to Reading on a golden winter afternoon is a very fine train ride
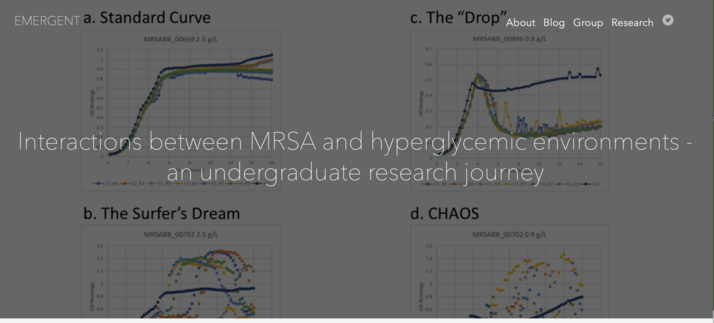
New lab blog post written by undergraduate researcher Alex Banul describing his project examining strain-level variation in MRSA growth in high sugar conditions. Alex showed an amazing level of maturity and poise to pull off both the project and the blog post. I feel he should submit this work as a short publication somewhere.
#MRSA #Staphylococcus #diabetes https://emergent.emory.edu/blog/posts/alexbog-feb2024/Interactions between MRSA and hyperglycemic environments - an undergraduate research journey
Introduction: A Texan’s First Summer in Atlanta This past summer and over the past year, I have had the opportunity to work in the Read lab as an undergraduate researcher. I am a recent graduate of the Emory College of Arts and Sciences where I received a B.S. in Biology and Philosophy. I came to Emory with the hope of participating in undergraduate research, and at the beginning of my junior year I had that hope realized when I took course IBS499R.
New blog post with @vishnu_raghuram "Diving into the pool: genome based strategies to effectively sample pathogen diversity", where we discuss our recent Mirobial Genomics paper. https://www.microbiologyresearch.org/content/journal/mgen/10.1099/mgen.0.001111
When is it best to sample single bacterial genomes versus pooled colony sweeps?
#bioinformatics #mrsa
https://emergent.emory.edu/blog/posts/vishnu_mgen_2024/


Comparison of genomic diversity between single and pooled Staphylococcus aureus colonies isolated from human colonization cultures
The most common approach to sampling the bacterial populations within an infected or colonized host is to sequence genomes from a single colony obtained from a culture plate. However, it is recognized that this method does not capture the genetic diversity in the population. Sequencing a mixture of several colonies (pool-seq) is a better approach to detect population heterogeneity, but it is more complex to analyse due to different types of heterogeneity, such as within-clone polymorphisms, multi-strain mixtures, multi-species mixtures and contamination. Here, we compared 8 single-colony isolates (singles) and pool-seq on a set of 2286
Staphylococcus aureus
culture samples to identify features that can distinguish pure samples, samples undergoing intraclonal variation and mixed strain samples. The samples were obtained by swabbing 3 body sites on 85 human participants quarterly for a year, who initially presented with a methicillin-resistant
S. aureus
skin and soft-tissue infection (SSTI). We compared parameters such as sequence quality, contamination, allele frequency, nucleotide diversity and pangenome diversity in each pool to those for the corresponding singles. Comparing singles from the same culture plate, we found that 18% of sample collections contained mixtures of multiple multilocus sequence types (MLSTs or STs). We showed that pool-seq data alone could predict the presence of multi-ST populations with 95% accuracy. We also showed that pool-seq could be used to estimate the number of intra-clonal polymorphic sites in the population. Additionally, we found that the pool may contain clinically relevant genes such as antimicrobial resistance markers that may be missed when only examining singles. These results highlight the potential advantage of analysing genome sequences of total populations obtained from clinical cultures rather than single colonies.
Emory University .. that's a major research institution with lots going on .. think I'll check their twitter page to see whats happening today .... oh
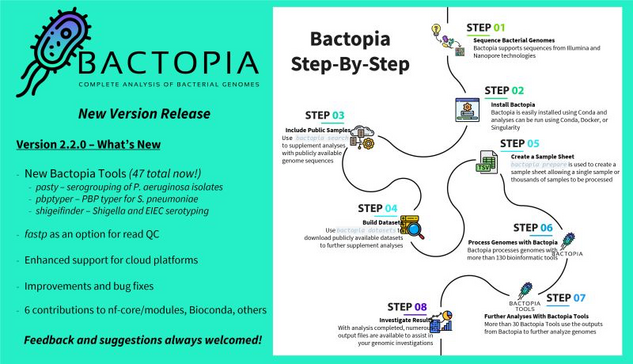
Bactopia v2.2.0 just released by @rpetit3
Fully featured pipeline for #bacteria #genomes analysis using #nextflow. More specific-specific tools for querying #pathogens! Better cloud support!
#bioinformatics #software