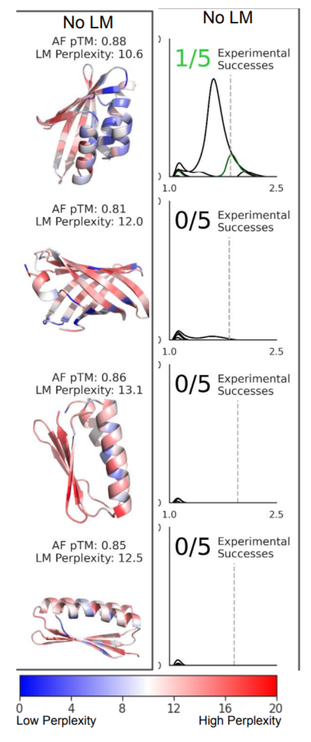
Efficiently generate de novo proteins by
- optimizing residue logits for max AF confidence
- redesigning the sequence using ProteinMPNN
Tested in the lab, including CryoEM structures
@chrisfrank662 @AKhoshouei @sokrypton @hendrik_dietz
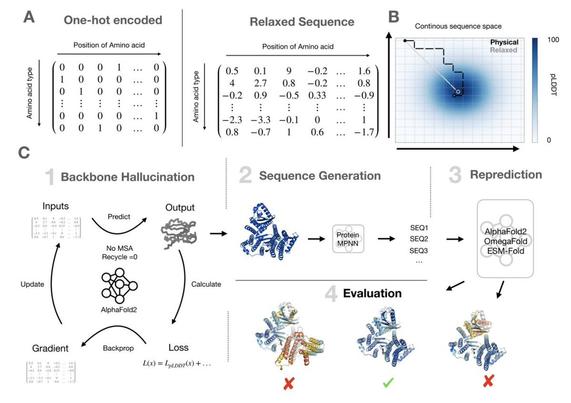

Efficiently generate de novo proteins by
- optimizing residue logits for max AF confidence
- redesigning the sequence using ProteinMPNN
Tested in the lab, including CryoEM structures
@chrisfrank662 @AKhoshouei @sokrypton @hendrik_dietz
@neuropunk
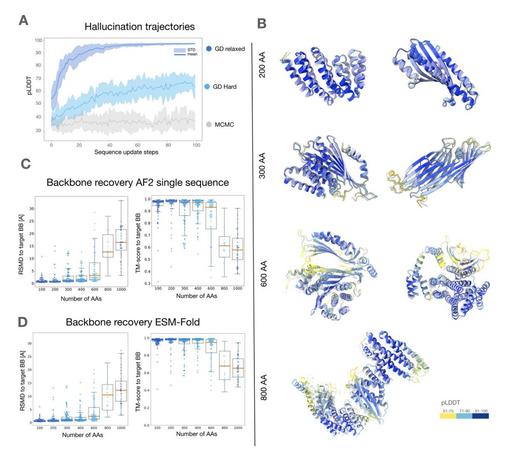
So now, during design, as soon as you get the desired structure, there is no longer any signal to update your sequence.
In this case, we wanted the structure to be fully encoded in the LM's contacts, and to avoid a situation where a more complex structure module starts hallucinating or improvising. (2/2)
@neuropunk
It's good for the structure prediction task, you want the model to be robust and recognize the bare minimum signal from the input sequence. But not good for the design task.
Let's say you have a suboptimal sequence that only partly encodes the desired structure. If your model is "too good", it will fill in the rest of the structure. (1/2)
check out the preprint:
https://www.biorxiv.org/content/10.1101/2022.12.21.521521v1
Thanks to all the amazing collaborators!
@robert_verkuil
@OriKabeli
@du_yilun
@BasileWicky
@LFMilles
@JustasDauparas
David Baker
@UWproteindesign
@TomSercu
@alexrives