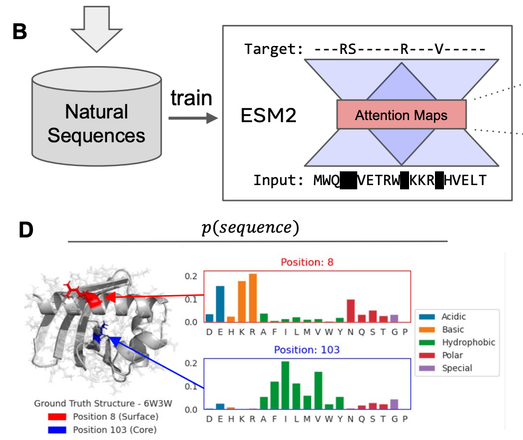
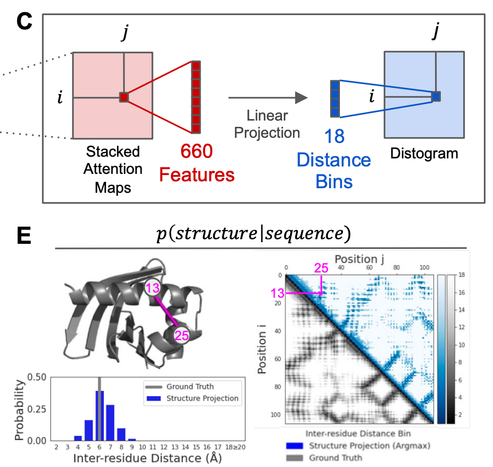
One issue with using methods like AlphaFold or RoseTTAFold for design is that they were trained to model P(structure|sequence). They were only trained on valid sequences. So if you use AF for design, you are likely to find adversarial sequences that trick the model. So you need a way to model the validity of the sequence or the P(sequence). Enter protein language models! (1/5)