Efficiently generate de novo proteins by
- optimizing residue logits for max AF confidence
- redesigning the sequence using ProteinMPNN
Tested in the lab, including CryoEM structures
@chrisfrank662 @AKhoshouei @sokrypton @hendrik_dietz
@sokrypton

Efficiently generate de novo proteins by
- optimizing residue logits for max AF confidence
- redesigning the sequence using ProteinMPNN
Tested in the lab, including CryoEM structures
@chrisfrank662 @AKhoshouei @sokrypton @hendrik_dietz
check out the preprint:
https://www.biorxiv.org/content/10.1101/2022.12.21.521521v1
Thanks to all the amazing collaborators!
@robert_verkuil
@OriKabeli
@du_yilun
@BasileWicky
@LFMilles
@JustasDauparas
David Baker
@UWproteindesign
@TomSercu
@alexrives

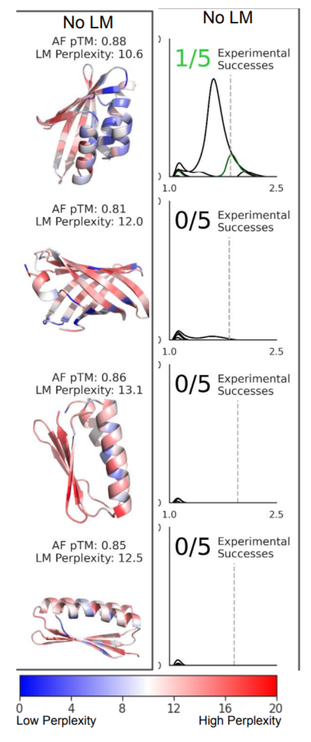
Now we can invert this model to find a sequence that matches a given backbone. (In this case, denovo designed backbones were selected, and any sequences remotely similar to the designed sequences were purged from the LM training set.)
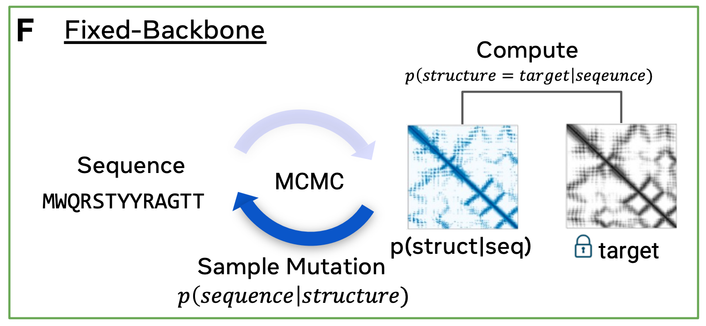
Given Bayes' theorem, by optimizing both the p(xyz|seq) and p(seq), we are also optimizing p(seq|xyz), since p(xyz) is constant. (3/5)
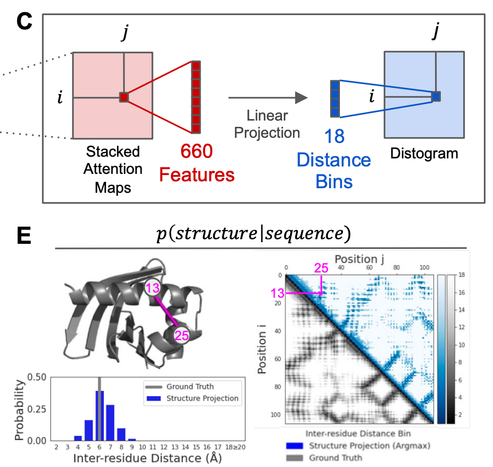
Given the observation that attention maps in the LMs correspond to contacts. One can train a linear projection from the attention maps to a distogram, allowing the modeling of P(structure | sequence). (2/5)
Papers showing LMs learn contacts:
https://arxiv.org/abs/2006.15222
https://www.biorxiv.org/content/10.1101/2020.12.15.422761v1
https://www.biorxiv.org/content/10.1101/2020.12.21.423882v2
Transformer architectures have proven to learn useful representations for protein classification and generation tasks. However, these representations present challenges in interpretability. In this work, we demonstrate a set of methods for analyzing protein Transformer models through the lens of attention. We show that attention: (1) captures the folding structure of proteins, connecting amino acids that are far apart in the underlying sequence, but spatially close in the three-dimensional structure, (2) targets binding sites, a key functional component of proteins, and (3) focuses on progressively more complex biophysical properties with increasing layer depth. We find this behavior to be consistent across three Transformer architectures (BERT, ALBERT, XLNet) and two distinct protein datasets. We also present a three-dimensional visualization of the interaction between attention and protein structure. Code for visualization and analysis is available at https://github.com/salesforce/provis.