New AlphaFold model quality estimator from @sokrypton does not require sequence alignments/ MSA. https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.129.238101
| Google Scholar | https://scholar.google.com.au/citations?user=Uc8nSqYAAAAJ&hl=en |

| Google Scholar | https://scholar.google.com.au/citations?user=Uc8nSqYAAAAJ&hl=en |
As always, large scale XLMS is intensly rich data & we have lots more stories (tech & bio) learned from Jason's amazing dataset. We are very excited for the future where the deluge of XL-MS datasets routinely inform the deluge of structure and interactome predictions!
Thank you to everyone involved @SydneyChemistry, @UNSWBABS, @SydneySOLES including @PayneResearch, @JoelPMackay, @XabiVC, Marc Wilkins, Clement Luong, Alex Norman & especially @jasonkklow! We welcome any feedback & feel free to reach out, incl. at #HUPO2022 IRL!
P.S. this is my first time using Mastodon, so sorry if o did if wrong and clogged up your feed!
Using the XLs to assess these predicted interfaces (following some QC - clashscore, pLDDT, interface size) revealed a bimodal distrubution of XL satisfaction (different to oberseved for monomers). Our XLs experimentally confirm structural interfaces of 167 dimeric PPIs.
Again, some models sig. expanded existing structures, docked binary PPIs known from previous interactomics studies, or both defined & localised a functionally predicted PPI for the first time! A newly validated interface was even near a disease-associated mutation.
XLd PPIs could also represent binary PPIs captured in larger multimeric complexes. We show how the XLs can be used to contextualise and inform the modelling of larger complexes to intelligently piece together the overall complex topology.
But our XLs also capture identities of + localise interfaces in >2000 binary PPIs! Many of these were known from previous high throughput #interactomics, but others were predicted, or a hop & skip away. Again - these are PPIs in native seqs, subcell locales, PTMs, cofactors.
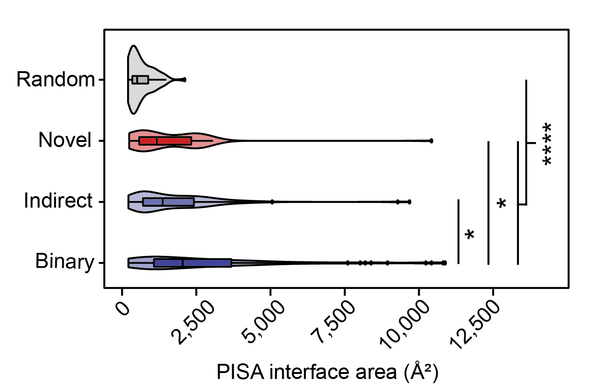
Using #ColabFold, Xabier Vazquez Campos submitted >500 of our XLd PPIs + 400 random pairs to Multimer-v2 modelling. All types of XLd PPIs had larger predicted interfaces than those for random pairs. We also found diferences in interfaces for PPIs detected by diff interactome techniques.

The advent of AlphaFold has resulted in a significant expansion of the predicted structural proteome, but it remains to be seen whether these predictions (trained on largely on in vitro structures) reflect bona fide biological structural conformations.
We found that XLs mapped to AlphaFold predictions for proteins w/o any experimental PDB structures actually performed BETTER than those falling within PDB-resolved regions! XLs in unresolved regions within PDB-resolved proteins performed worse (eg. conformational dynamics, disorder).
The XLs were super useful in assessment of AlphaFold predictions - we were able to confirm the regions within expanded existing structures, homologous proteins & provide 1st ever experimental validation for >600 entirely novel structures! XLs also identify quirks in models, and can be theoretically used to guide model refinement.
Mapping to ~10000 experimental structures curated into the PDB = vast majority of in vitro structures now validated by our XLs - notably captured in proteins w/ native seqs, abundances, PTMs & cofactors. Those that differed usually represented interplay b/w structure & PTMs or complexes.
But most XLs could NOT be mapped to the PDB as most proteins are yet to be studied by low throughput but high resolution structural biology tools. Others are simply recalitrant to purification and/or crystallization.
