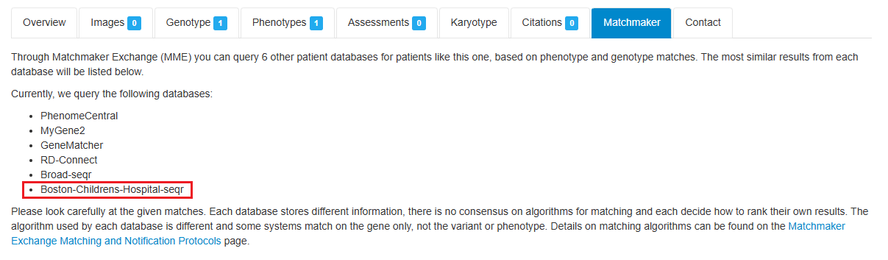
DECIPHER users can now search for patient matches in Boston Children’s Hospital seqr in addition to PhenomeCentral, Broad seqr, GeneMatcher, RD-connect and MyGene2 using Matchmaker Exchange
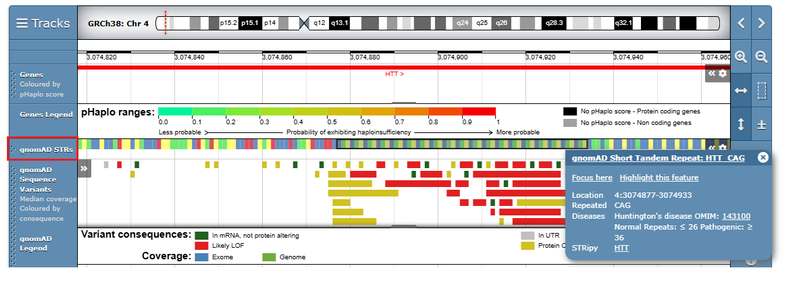
A gnomAD Short Tandem Repeat track is now available on the genome browser which displays information about 60 disease associated repeat loci. The associated diseases are displayed along with the normal and pathogenic repeat lengths, and links to STRipy
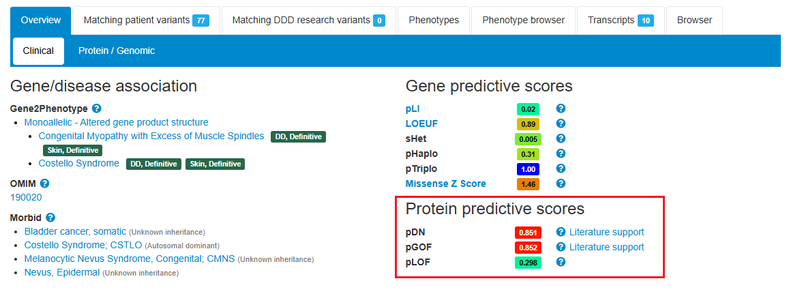
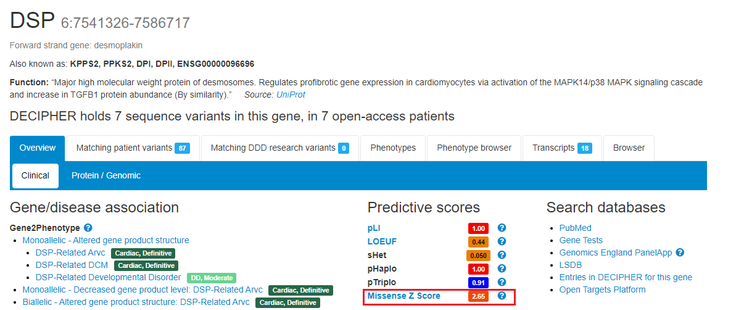
Protein predictive scores which predict the likelihood that the protein is associated with a dominant-negative, gain-of-function or loss-of-function mechanism are displayed. Curated literature support for a molecular disease mechanism is also shown
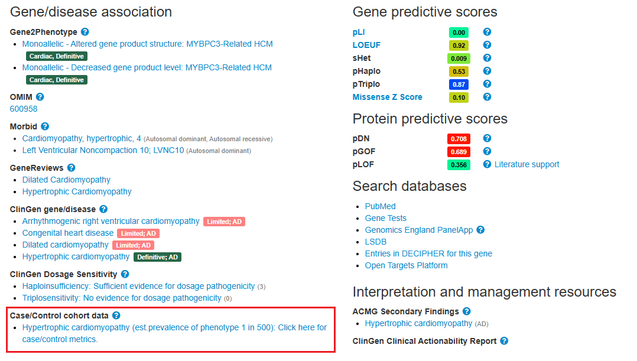
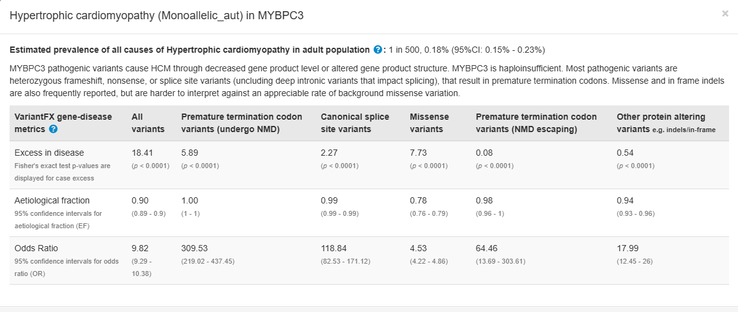
The prevalence for cardiomyopathies is also now displayed for genes associated with cardiac disorders
Cardiac case/control cohort data, which demonstrates the confidence of cardiac gene-phenotype relationships associated with specific variant classes, has been updated; more variant classes e.g. canonical splice site variants
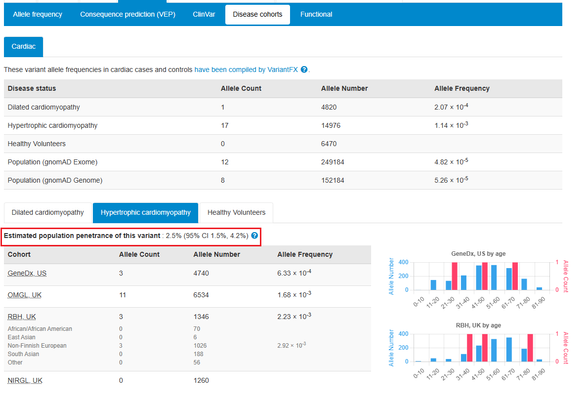
Estimated population penetrance for variants associated with cardiomyopathies are now displayed alongside cardiac allele frequencies. This information is useful when considering secondary findings.
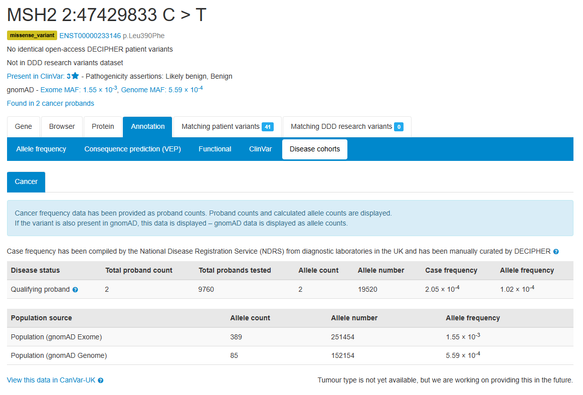
Cancer case frequency data compiled by the National Disease Registration Service from diagnostic laboratories in England is now displayed. Nearly 4,500 variants in 13 cancer susceptibility genes including BRCA1, BRCA2, MSH2, PTEN and SMAD4.
DECIPHER version 11.29 has been released. See the new features at
https://www.deciphergenomics.org #variantinterpretationDECIPHER v11.29: Mapping the clinical genome
DECIPHER helps the clinical community share and compare human genome variants and phenotypes in a database of tens of thousands of patients worldwide
Happy 20th Birthday DECIPHER
The DECIPHER team celebrating the platform facilitating variant classification, patient diagnosis and empowering rare disease research for 20 years.
Missense Z scores based on gnomad v4.1, are now available on gene pages. These scores measure the amount that a gene is constrained for missense variants.