Tootorial on the recent preprint on inferring ARGs for SARS-Cov-2:
The method is essentially very simple. We incrementally add batches of samples to the ARG, using the Li and Stephens HMM to find likely attachment points (or paths, if recombination is detected), resolve clusters using standard tree methods, and finally some parsimony tweaks.
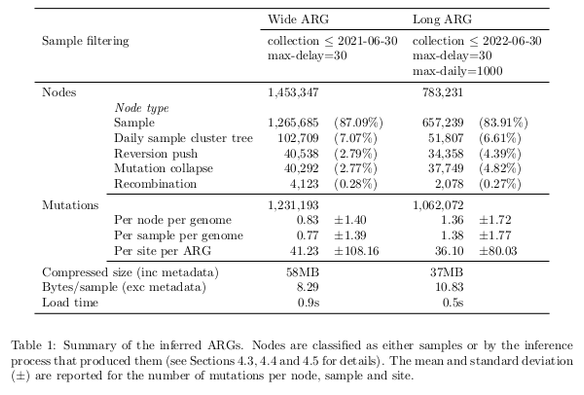
We infer two large ARGs: the "Wide" (1.27M samples, up to mid 2021) and "Long" (657K samples, up to mid 2022). Both are very concise and efficient to work with - whole SARS-Cov-2 genomes are stored in 8.29 **bytes** on average in the Wide ARG!
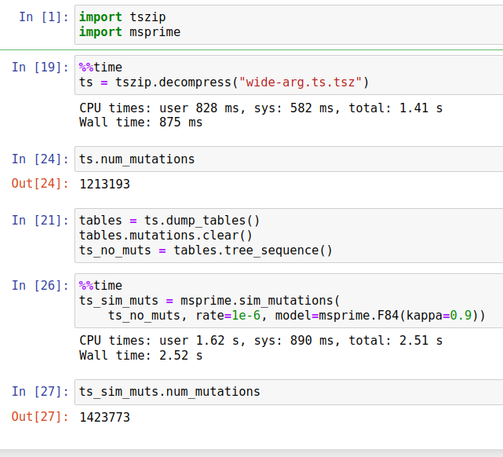
We can analyse these ARGs using the well-established and feature-rich #tskit library (and surrounding ecosystem). All of the analyses for the preprint were done using Jupyter notebooks, and most run in seconds on a standard laptop.
Building on top of this existing library and package infrastructure has huge advantages. For example, here's some code where we load the Wide ARG, and simulate 1.4 million mutations under the Felsenstein 84 model using #msprime (params arbitrary). This takes 2.5 seconds.
The sc2ts method itself is a thin wrapper around existing high performance components. The actual inference code is only about 1300 lines of Python. We can infer viral ARGs at this unprecedented scale with no compiled code **at all**.
https://github.com/jeromekelleher/sc2ts/blob/main/sc2ts/inference.py

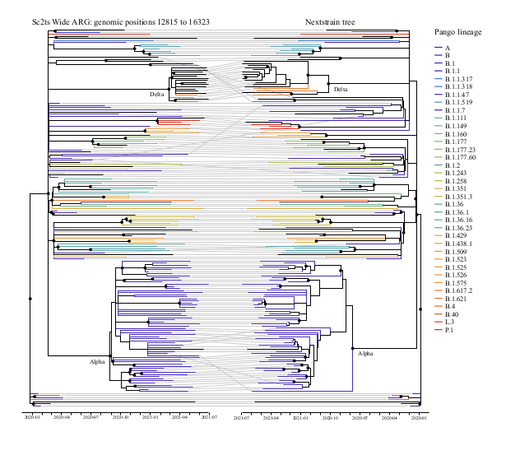
The method works quite well, despite the simplicity of the tree building model and reliance on parsimony heuristics. Here is a comparison of a "backbone" phylogeny compared to a Nextstrain tree. While there is clearly room for improvement, we're mostly doing well.
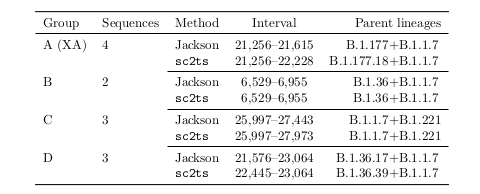
The whole point of an ARG is to account for recombination though - so how do we do there? As far as we can tell, very well. For example, we compare with the early recombinants detected by Jackson et al, with near perfect agreement:
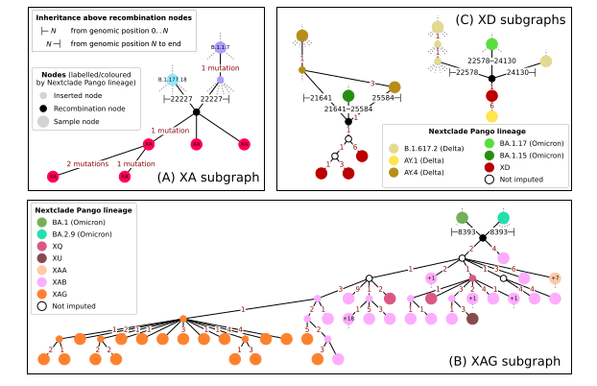
We can characterise the origin of recombinants very precisely. Here are some subgraphs showing the origins of Pango X lineages and clustering of samples (based on Nextclade Pango lineage assignments).
For this initial version we have focused on validating the method, and identifying areas for improvement. Overall, we see excellent agreement with established results, which gives a lot of confidence in the overall strategy.
With some further development of the method, the inferred ARGs could be a hugely powerful platform for subsequent analysis, allowing ongoing recombination to be *fully accounted for* in phylogenetic analyses of SARS-CoV-2, in an efficient and convenient computational platform.
If you're interested in using these ARGs for your analyses, or you think you can help improve the method, please get in touch! With some relatively minor improvements we should be able to scale up to >10M samples, leading to truly pandemic scale ARGs.
ps. See also the summary post on virological:
https://virological.org/t/towards-pandemic-scale-ancestral-recombination-graphs-of-sars-cov-2/936

Towards Pandemic-Scale Ancestral Recombination Graphs of SARS-CoV-2
Towards Pandemic-Scale Ancestral Recombination Graphs of SARS-CoV-2 Shing H. Zhan1, Anastasia Ignatieva2,3,*, Yan Wong1,*, Katherine Eaton4, Benjamin Jeffery1, Duncan S. Palmer1, Carmen L. Murall4, Sarah P. Otto5, Jerome Kelleher1,† 1Big Data Institute , Li Ka Shing Centre for Health Information and Discovery, University of Oxford, United Kingdom 2Department of Statistics, University of Oxford, United Kingdom 3School of Mathematics and Statistics, University of Glasgow, United Kingdom 4Nati...