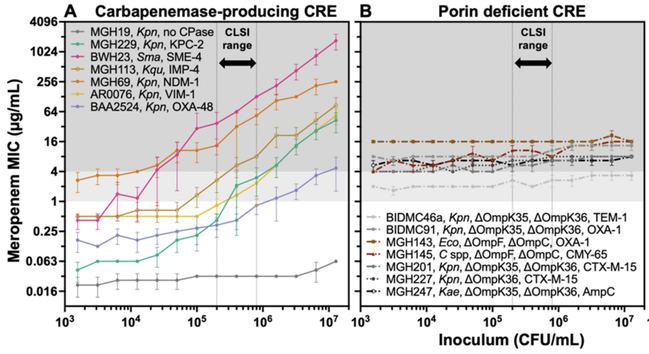
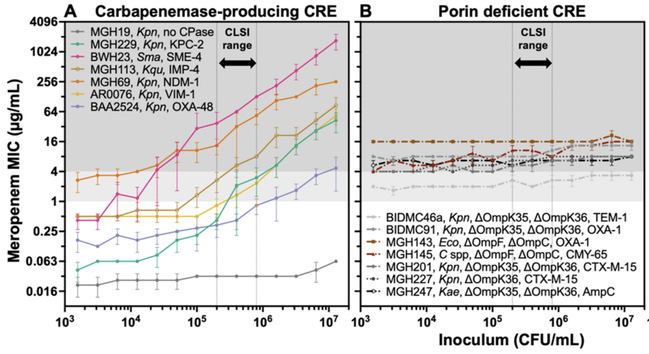
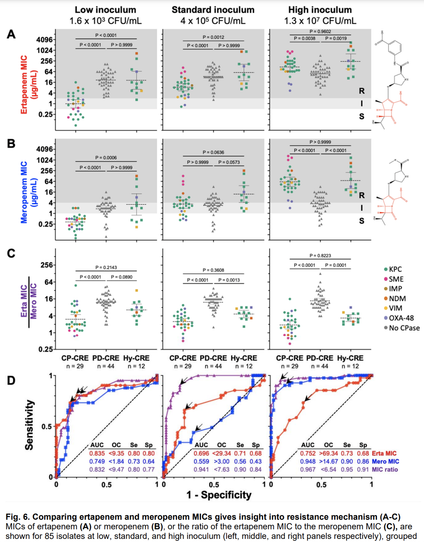
Until a project led by my lab's first postdoc, Alex Jaramillo, & stellar RA Kyra Taylor, my thoughts on the inoculum effect (IE) - the more bacteria there are, the greater the conc of antibiotic required to kill them - were fuzzy.
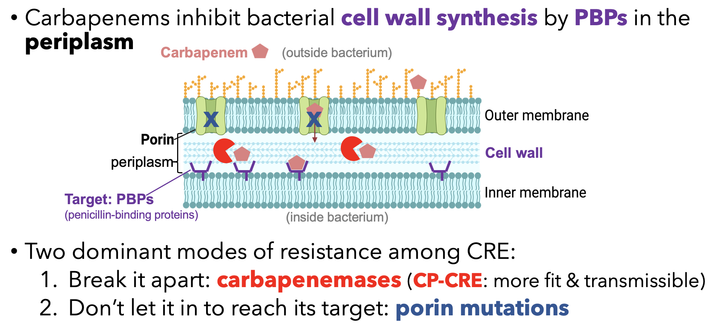
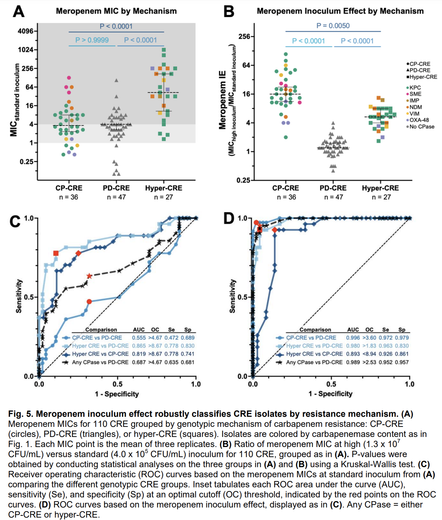
In a new preprint, imo we clarified the carbapenem IE: carbapenemases act as shared goods for bacterial communities. All carbapenemase-producing strains had a strong IE; those resistant due to porin deficiency had no IE at all.
This has key consequences…
🧵 1/n